




Scientific highlights in polymer science
Here you will find selected scientific highlights of the collaborative research center in the fields of polymer physics, polymer chemistry and biophysics. Our transregional research center is a joint initiative of Martin Luther University Halle-Wittenberg and Leipzig University.
Scientific highlights of SFB/TRR 102
Aggregation of biological macromolecules
Highlights 2023
Lipid oxidation controls peptide self-assembly near membranes through a surface attraction mechanism
Despite considerable advances in research, there is still no cure for many age-related diseases, such as Alzheimer’s. One hypothesis for the development of Alzheimer’s is the aggregation of peptides, i.e. their “clumping”, in the brains of people with the disease. But stress has also been suspected as a possible trigger. Scientists from Leipzig University, Monash University in Australia, the Leibniz Institute of Surface Engineering (IOM) and the University of Göttingen have investigated the links between oxidised cell membranes and peptide aggregation and have now published their findings in the journal Chemical Science.
Highlights 2022
Hydrodynamic Manipulation of Nano-Objects by Optically Induced Thermo-Osmotic Flows
The manipulation, control and transport of molecules and particles in fluids is a key feature of microfluidics for all kinds of applications ranging from macromolecule analysis to chemical synthesis. Fluids in small channels are thereby driven by macroscopic pressure gradients, surface acoustic waves or electro-osmotic flows. We have developed a method that generates strong currents in small liquid volumes by remote heating of a thin metal film at a liquid-solid interface. The flows are created by van der Waals forces between water and the metal, which also facilitate nanoparticle trapping by interfacial forces. The dynamic variation of the temperature fields enables the generation of complex flow patterns that, together with other thermal effects also provides new ways to handle peptides in protein aggregation studies.
Highlights 2021
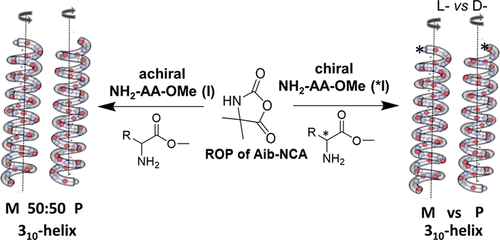
Chiral amines as initiators for ROP and their chiral induction on poly(2-aminoisobutyric acid) chains

Chirality is a unique characteristic of living systems, as an example represented as either left- or right-handed helical conformations with an outstanding impact to self-assemble into higher-order structures. α-Aminoisobutyric acid (Aib) is an achiral amino acids and known for its helical building properties, leading to either left or right-handed helices in poly(peptides). We have prepared poly(Aib)s by ring opening polymerization and investigated their preference to organize into either left- or right-handed helices by choice of a chiral inductor, placed at the head of the polymer chain. By using chiral amines as initiators for the polymerization reaction, we are able to induce a chirality on the poly(Aib) chain and can select a certain handedness helix by the choice of the initiator. As a major finding we could prove that chirality is induced only by the sterical influence of the initiator, preferentially via hydrogen bonds. The here investigated system thus allows to amplify chirality by only minor amounts of a chiral inductor, being potentially useful for spin-selection, chemical reactions or the organization of the helices into superstructures.
Highlights 2020
Chirality Control of Screw-Sense in Aib-Polymers: Synthesis and Helicity of Amino Acid Functionalized Polymers
2-Aminoisobutyric acid (Aib) is an essential amino acid, leading to the formation of peptAibols as microbiologically active peptides and proteins. The formation of helices is crucial for their function, also mediating folding of proteins when present as several crowded units within a longer peptide-stretch. Here, we report on the preparation of disctinct Aib-polymers with ring-opening polymerization. We systematically investigated the helical screwsense of the Aib-polymers and induced either left- or right-handed screw senses by adding chiral amino acids. Moreover, we were able to switch the chirality of the polymer with light-induced triggers. The here shown transfer of chirality over large distances is exemplary for cooperative behavior in biologically active peptides and proteins, wherein Aib is a functional constituent. The so formed transient helices are highly dynamic, representing an important model system for protein folding and protein aggregation.
β-Turn mimetic synthetic peptides as amyloid-β aggregation inhibitors
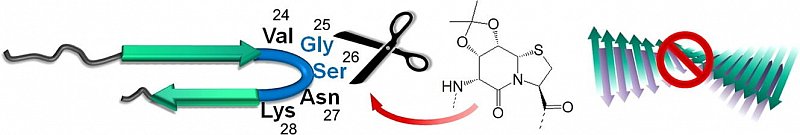
Aggregation of amyloid peptides results in severe neurodegenerative diseases. While the fibril structures of Aβ40 and Aβ42 have been described recently, resolution of the aggregation pathway and evaluation of potent inhibitors still remains elusive, in particular in view of the hairpin-region of Aβ40. We here report the preparation of artificially modified Aβ40-proteins containing synthetic turn mimetic structures, replacing 2 amino acids in the turn-region. In these model systems, reduced fibrillation is observed with conformationally constrained turns, while conformationally flexible beta-turn-mimetics lead to enhanced fibrillation, highlighting the underlying beta-turn as a contraining folding unit to mediate subsequent fibrillation.
Highlights 2019
Synthesis and Mechanochemical Activity of Peptide-Based Cu(I) Bis(N-Heterocyclic Carbene) Complexes
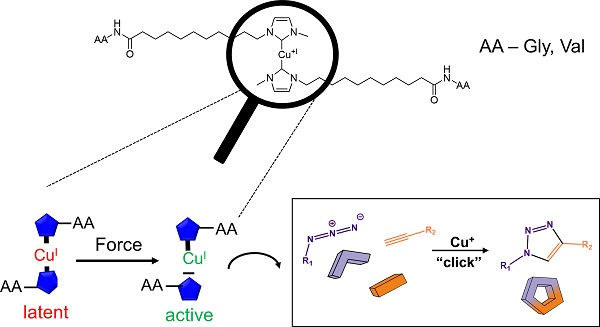
With the class of shock-absorbing proteins, nature created some of the most robust materials combining both mechanical strength and elasticity. The engineering of their micro- and nanostructure allows for a defined ordering of these highly complex, strongly hydrated gels. Measuring and quantifying local stress within such materials is crucial in further optimizing their material properties. Here, we report on the embedding of an internal stress-sensor into such a material, which allows quantification of nanoscaled stress. To this effect we report on the synthesis of two complexes and show their mechanochemical activation by ultrasound.
Thermophoretic trap for single amyloid fibril and protein aggregation studies
A major difficulty in the investigation of protein aggregation and other macromolecular nucleation and growth processes is the heterogeneity of the ensemble studied, which prevents the direct identification of the contributing processes. Using a sophisticated technique of thermophoretic trapping, we provide a method that eliminates this difficulty by confining individual amyloid fibrils in liquids without any surface attachment. With the help of this technique, we are able to follow the growth process of protein fibrils over time periods of hours allowing the observation of rare events like fibril fracture.
Highlights 2018
Impact of nanoparticles on amyloid peptide and protein aggregation
The effect of surfaces on amyloid formation is investigated in project B01. By a combination of experimental investigations of fibrilliation kinetics and molecular dynamics simulations it was possible to elucidate the effects of metallic nano particles on fibril formation of selected amyloid-forming sequences. The results indicate that a thin protein layer adsorbed on the metal surface, the so-called corona, is the key to understand the observed effects. It depends on the internal structure of the corona, if fibrillation is nucleated at the surface and therefore enhanced.
- T. John, A. Gladytz, C. Kubeil, L. L. Martin, H. J. Risselada, and B. Abel, Nanoscale 10, 20894 (2018).
See also
Highlights 2017
Perturbation of the F19-L34 Contact in Amyloid β (1-40) Fibrils Induces Only Local Structural Changes but Abolishes Cytotoxicity
The effects of specific local mutations on the local and global structure of Aβ (1-40) peptides are in the focus of the investigations in project A06. NMR experiments and computer simulations were combined in a collaboration together with A09 to explore structural details of fibrils formed by one specifically selected mutation of an Amyloid β (1-40) peptide. Although morphologically robust, local perturbations of the sequence led to moderate structural alterations on a local scale with large impact on the physiological properties, as the modified system showed no toxicity. Similarly strong effects on toxicity accompanied by changes in fibrillation kinetics were found for other local mutations.
Foundation of the Outstanding Toughness in Biomimetic and Natural Spider Silk
Spider dragline silk exhibits the highest toughness among all known materials. Even though their morphology has been thoroughly studied, it was not possible to recreate comparable materials until recently. On the basis of fibers from the recombinant protein (AQ)12NR3 –which are so far the only threads able to compete in toughness with natural silk– the molecular origin of the fibers’ high toughness is elucidated. It is demonstrated that in addition to the sample’s morphology its functional structure is essential for the material’s performance; post-spinning strain orients protein chains and induces a microscopic non-equilibrium. As a consequence macroscopic stress is directly transduced to crystalline fiber segments dissipating mechanical load. This mechanism known from natural spider silk is for the first time revealed for artificial silk.
Highlights 2016
Coupling and Decoupling of Rotational and Translational Diffusion of Proteins under Crowding Conditions
Cataract, that is, the loss of vision due to growing opacity of the eye lens, is one of the most frequent deseases with a correspondingly huge social (and economic) impact on humankind. It is associated with either phase separation or denaturation and ensuing aggregation in the concentrated solution of various lens proteins, the so-called crystallins. Our understanding of the molecular underpinnings of these processes is as yet incomplete, and it is one of the goals of project A08 to test in how far amyloid formation may be relevant in this context. As a step towards this goal, the constrained diffusive motion of proteins in such a highly crowded environment should be characterized first. It is well established that the large-scale translational diffusion is governed by the viscosity of the system, in agreement with the Stokes-Einstein relation. However, the applicability of the coresponding Stokes-Einstein-Debye relation for rotational diffusion, which is more localized short-time process, is still poorly understood. Using a combination of NMR and fluorescence spectroscopy techniques, we investigate the (de)coupling of the rotational from the translational diffusion in a series of crowded model proteins, revealing that proteins can span the whole range of phenomena in dependence of their complex mutual interactions.
Highlights 2015
Single Molecules Trapped by Dynamic Inhomogeneous Temperature Fields
Holding single molecules in liquids is still a challenge as Brownian motion fueled by thermal energy is randomizing molecular position quickly and common forces to counteract this erratic motion commonly scale with the volume. The benefits of hold and manipulating a single molecule are however huge as it would allow for long time observations of molecular conformation detecting rare events, biomolecular reactions and true bi-molecular interaction studies. Here we report on a method which employs the fuel of Brownian motion - thermal energy - to confine Brownian motion of single molecules in liquid. By generating time-dependent feedback controlled inhomogeneous temperature fields with a laser heated gold structure, we create thermophoretic drive fields which allow the confinement of single molecules in solution. The feedback control and the inhomogeneous character of the temperature field even allow for a trapping a well controlled number of multiple molecules. We expect that this simple method and an extension to large arrays of traps will pave the way for controlled molecular interactions studies.
See also
Highlights 2012
Solid-state NMR Reveals a Close Structural Relationship between Amyloid-β Protofibrils and Oligomers
Proteins, essential components of every living being, have been posing riddles to researchers worldwide for decades. The function of these long-chain biomolecules is determined by the spatial configuration. Strictly speaking a protein folds into a specific three-dimensional structure. Properly folded, the protein can fulfill the specific biological function in our body, e.g., the control of cell growth.
However, if anything is wrong with the folding - known as the misfolding of proteins - toxic substances can arise that might lead to diseases such as Alzheimer's disease and diabetes. Through intermediate stages, a gradual aggregation of misfolded proteins results in the so-called mature fibrils. This final stage possesses the form of elongated rods which are composed of helically wound, zipper-like interconnected strips.
Holger Scheidt, from the group of Daniel Huster at University of Leipzig, and his colleagues have now succeeded in finding a greater similarity of the internal three-dimensional structure of the two intermediates for the first time. Mature fibrils, however, show an altered spatial arrangement. In detail, the protein amyloid-beta (1-40) was examined by using high-resolution nuclear magnetic resonance (NMR) spectroscopy within the CRC project A06.
Crystallization of synthetic polymers
Highlights 2023
How entanglements determine the morphology of semicrystalline polymers
The majority of polymers used in the solid state as construction or packaging materials are semicrystalline. Their typical microscopic structure consists of nanoscopically thin yet stiff crystalline layers separated by rubbery amorphous layers. This specific morphology is responsible for the advantageous mechanical properties of these materials. Decisive parameters are the thicknesses of the crystalline and amorphous layers that together determine the crystallinity and the entanglement concentration in the amorphous layers. While the factors determining the crystal thickness are well investigated, there is a lack of understanding for the amorphous layers. Using a model polymer for which the entanglements are retained during crystallization, we show that their thickness is controlled by the entanglement concentration that attains a specific maximum value.
Highlights 2022
Competition between crystal growth and intracrystalline chain diffusion determines the lamellar thickness in semicrystalline polymers
The characteristic morphological feature of semicrystalline polymers crystallized from the melt is a nanoscopic two-phase structure of thin lamellar crystals separated by disordered amorphous layers. This morphology is largely responsible for the favorable mechanical properties of semicrystalline polymers.
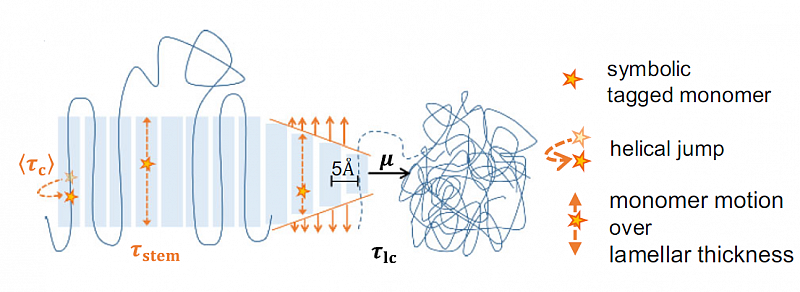
Our work addresses a classical question in polymer physics: which factors control the thickness of the crystalline layers? By combining a variety of complementary experimental techniques, especially small-angle X-ray scattering and NMR methods, we came to a consistent understanding that highlights the role of intracrystalline mobility, a typical feature of many semicrystalline polymers. The semicrystalline morphology is determined by the competition between crystal growth (characterized by the timescale of stem attachment) and simultaneous thickening of the crystallites (timescale of the intracrystalline chain diffusion as a measure for the mobility of the chains within the crystal). The results explain not only the strong temperature dependence of the crystal thickness found for some polymers but also the large variation of this quantity among different polymers affecting the degree of crystallinity and by that the mechanical properties of the material.
Highlights 2021
Microscopic characterization of poly(sulfur nitride)

Poly(sulfur nitride) (SNx) is a unique synthetic polymer as it has the conductivity of metals at room temperature and is the only synthetic polymeric superconductor. In this work, bulk crystals as well as thin films of SNx have been synthesized and investigated. The bulk crystals consist of fibers, which show a microscopic macroscopic twinning. The crystallographic orientation in thin SNx-films was studied by Grazing Incidence Wide Angle X-ray Scattering (GI WAXS). Conductive Atomic Force Microscopy on thin films provides information on the electrical conductivity of SNx crystal together with its morphology in the nm range. The current-voltage (I-V) curves show ohmic behavior indicating the metallic nature of SNx.
Molecular heterogeneities in the thermal expansivity of polyalcohols
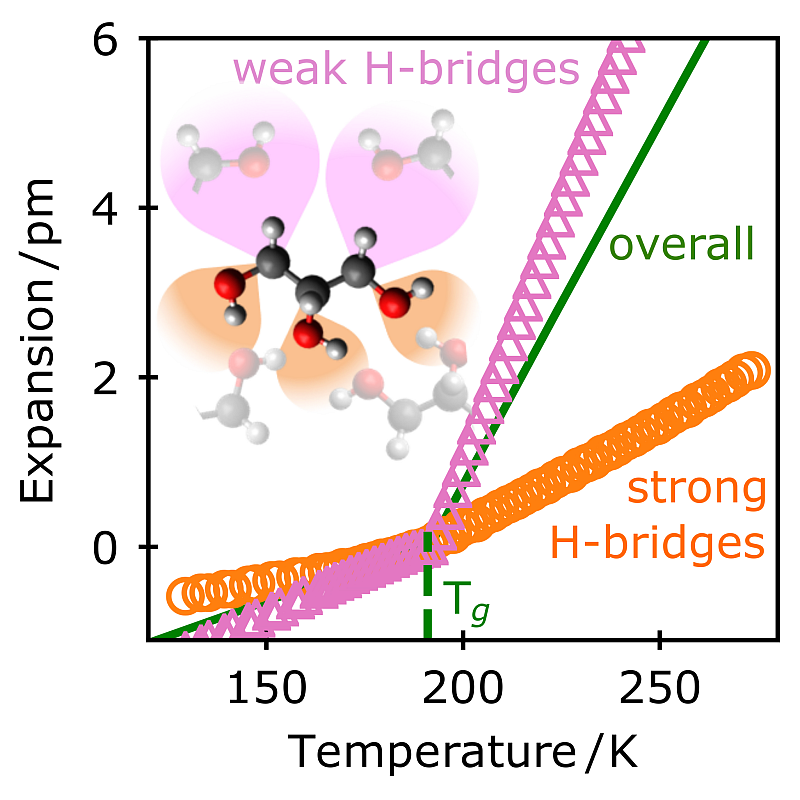
Density is the key quantity for nearly all the numerous theories of the (dynamic) glass transition of supercooled liquids and melts. As a mean field quantity, it is used to describe correlations and heterogeneities between regions consisting of several molecules. In contrast, the question how density is created by the interactions (i.e., bonds) within a molecule and to its nearest neighbors is almost unexplored. To investigate this for example of a homologous series of polyalcohols (glycerol, threitol, xylitol, and sorbitol), Fourier-Transform InfraRed (FTIR) spectroscopy is carried out in a wide range of temperatures from far above to far below the calorimetric glass transition Tg. This enables us to determine the potentials and hence the bond lengths of specific intramolecular and intermolecular interactions. While the former has an expansion coefficient of (∼0.1 pm/100 K) with only smooth changes, the latter shows a 30–40 times stronger response with pronounced kinks at Tg. A comparison with the overall expansion based on mass density reveals that one has to separate between strong (OH⋅⋅⋅O) and weak (CH⋅⋅⋅O) intermolecular hydrogen (H)-bridges. Despite the fact that the latter dominates glassy dynamics, their expansivity is 5 times smaller than that of the weak H-bridges. It is to be expected that such heterogeneities on intramolecular and intermolecular scales are a general phenomenon in liquids and glassy systems demonstrating especially the necessity of atomistic simulations.
Interfacial Dynamics in Supported Ultrathin Polymer Films—From the Solid to the Free Interface
In many day-to-day applications, thin layers of polymers are placed in close contact with solid substrates, thereby amplifying the role of interfaces in determining the ensuing properties. Consequently, for close to three decades now, there has been concerted scientific effort toward gaining a fundamental understanding of how spatial confinement affects, inter alia, dynamics in polymers. In some of the experiments, an indirect approach is employed where probe molecules are placed in the polymer film and used as reporters of the host’s dynamics, a procedure that demands several nontrivial assumptions. However, for a nondestructive route, nanostructured electrodes for dielectric measurements provide direct access to molecular fluctuations in films having solid/polymer and polymer/air interfaces. Complemented by atomistic molecular simulations, it is now possible to quantitatively determine the extent of the interfacial regions that bear modified mobilities relative to bulk.
Highlights 2020
Independent Variation of Transition Temperature and Prefrozen Layer Thickness at the Prefreezing Transition
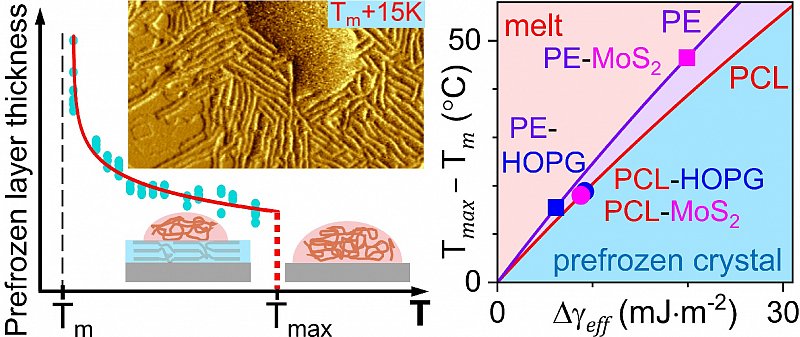
It is well-known that crystallization of liquids typically starts at the interface to a foreign solid surface. In general, interface-induce crystallization can take place either via heterogeneous nucleation or via prefreezing. Of special interest is the latter phenomenon, which refers to the abrupt formation of a mesoscopic and thermodynamically stable crystalline layer at the solid interface above the melting temperature of the material. Over the past few years, we carried out systematic investigations of prefreezing in a series of various polymer-substrate systems using direct in situ AFM and also proposed a phenomenological theory of prefreezing. In the current research work, we further extended our study of prefreezing to a new polymer-substrate system, poly(ε-caprolactone) (PCL) on a molybdenum disulfide (MoS2) substrate and compare the results with those of our previous studies of PCL prefrozen on graphite as well as polyethylene prefrozen on the same two substrates. In particular, we demonstrate that the transition temperature and minimum thickness of the prefrozen layer at the prefreezing transition can vary independently. Moreover, we also show that our diverse experimental findings on prefreezing of all four investigated systems can be consistently explained by the phenomenological theory. Our article provides the state of the art on the topic and unveils the exact role that the interfacial free energies play in controlling prefreezing at the melt-solid interface. It is worth mentioning that our results are not limited to polymer crystallization only but also valid for crystallization of other liquids on solid surfaces.
Tuning layered superstructures in precision polymers
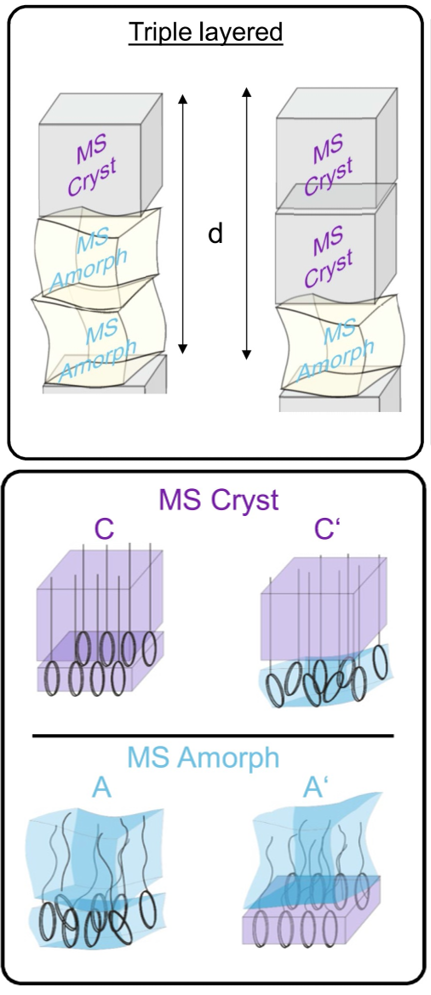
Precision polymers have become an important class of materials for various applications. Precise placement of functional groups at well-defined locations along a linear polyethylene backbone could introduce interesting properties due to the chemical nature and interactions between the groups. Most precision polymers are known to exhibit nano-layered states with typical periodicities in the 1-3 nanometer range.
The packing of the methylene sub-units within the layers depends upon the size and nature of the adjacent functional group as well as on the length of the methylene sequence giving rise to different polymorphic states. This addresses fundamental questions regarding the competition between the packing tendencies of the individual sub-units viz. functional groups and methylene sequences for structure formation processes in such complex systems. It is demonstrated that using diaminopyridine (DAP) group capable of introducing interactions via H-bonding or pi-stacking can stabilize different layered superstructures containing up to three monomeric repeating units along the chain axis. Varying the DAP to methylene units ratio (methylene spacer length) allows for tuning the nature of the layered structures. A rational design of precision polymers where the energetic contributions of methylene sequences and the functional groups compete can be used as possible strategies to tailor the long range ordered layered morphology.
Fingerprints of homogeneous nucleation and crystal growth in polyamide 66 as studied by combined infrared spectroscopy and fast scanning chip calorimetry
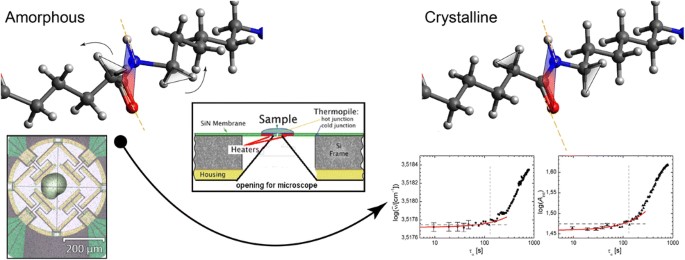
Polymers in general became an essential part of modern life with uncountable applications. The particular conditions during melt processing affect the resulting polymers macroscopic properties and it is therefore mandatory to pay attention to crystallization on the microscopic scale. Crystallization starts with an initial nucleation process within the supercooled sample followed by crystal growth. Thereby, nucleation can be either homogeneous with nuclei solely formed by polymer chains through thermodynamic driving forces or heterogeneous with nuclei established at interfaces. Since polymer crystallization is generally based on heterogeneous nucleation, it is mandatory to suppress heterogeneous nucleation in order to study homogenous nucleation in the bulk. This is only possible through sufficient fast cooling, and the first combined chip calorimetric-IR spectroscopic experiment is presented here on the example of PA66.
Highlights 2019
Phenomenological Theory of First-Order Prefreezing.
The microscopic ordering process that a liquid undergoes during crystallization is often initiated at an interface to a solid. Different processes have been suggested by theory to occur at this interface. A particularly interesting process is prefreezingthe formation of a thin crystalline layer at the interface already at temperatures above the melting temperature. A direct experimental observation of this process was only recently achieved. We here now present a phenomenological theory of prefreezing and analyze the thermodynamic properties of the prefrozen crystalline layer. Specifically our theory describes the first order nature of the transition and allows a quantitative analysis of previously obtained experimental data for poly(ε-caprolactone) crystallized on graphite via prefreezing. The validity of our results is not restricted to polymer systems but contributes to our general understanding of crystallization.
Highlights 2018
Crystallization in melts of short, semiflexible hard polymer chains: An interplay of entropies and dimensions.
What is the driving force for the crystallization of a melt of semiflexible polymers? This fundamental question has been addressed in project A07. Different from the case of crystallization of small molecules, which can be reduced in a most rudimentary model to the translational ordering of hard spheres, polymer crystallization always combines translational and orientational (conformational) ordering. Stochastic approximation Monte-Carlo simulations that give complete thermodynamic information were used to study an ensemble of short chains, for which kinetics do not yet play a role. The authors were able to show that in fact the orientational interaction sets the thermodynamic driving force whereas translational ordering follows downstream.
Crystallization of Poly(ethylene oxide) on the Surface of Aqueous Salt Solutions Studied by Grazing Incidence Wide-Angle X‐ray Scattering

Polymers under constraints in thin films have been studied intensively in recent years caused by their fundamental role for such sophisticated applications as lithography for electronics, surface engineering for biomedical devices, and sensor technology. During solvent evaporation, spin coated polymer films are frozen in a nonequilibrium state which leads to relaxation processes when semicrystalline polymers are kept above the melting temperature. Thus, after film formation of semicrystalline poly(ethylene oxide) (PEO) on silicon wafers, a pseudo dewetting mechanism of 50−100 nm thin polymer layers in the melt is followed by crystallization upon cooling. An approach to investigate thin layer crystallization is the use of Langmuir experiments. A polymer solution (e.g., in chloroform) is spread on an aqueous surface of a Langmuir trough between movable barriers. After solvent evaporation the barriers are compressed to decrease the available surface, that is, to continuously increase the polymer surface concentration. By this method, the polymer film can be varied from separated chains (i.e., gaseous phase) to a continuous layer with several nanometer thicknesses (i.e., liquid or crystalline phase), as long as the polymer does not dissolve in the subphase. Recent results from B07 demonstrated that PEO crystallizes on K2CO3 aqueous solution during compression on a Langmuir trough. PEO is usually considered as a water-soluble polymer because of strong interactions between ether oxygen and water, but it has also an amphiphilic character. By addition of kosmotropic salts like K2CO3 to the water subphase, dissolution of PEO is reduced and completely prevented at higher salt concentrations because of the Hofmeister effect. Using Brewster angle microscopy, the dendritic shape of the crystals is observed, which are floating free on the solution surface. With grazing-incidence wide-angle X-ray scattering (GIWAXS), the orientation of the crystallites has been be identified.
Interface-Induced Crystallization of Polycaprolactone on Graphite via First-Order Prewetting of the Crystalline Phase
The crystallization of liquids is often initiated at the interface to a solid. Due to the difficult accessibility of a buried liquid-solid interface, direct experimental observations of interface induced crystallization have been scarce and only a recently crystallization induced by prefreezing, which had been theoretically predicted beforehand was observed in experiment. In prefreezing, a thin crystalline film forms on a solid substrate in a finite temperature interval above the bulk melting temperature of the liquid. The thickness of this prefrozen films increases on approaching the melting temperature and continues to grow into the bulk upon further cooling.
The phenomenon can be understood in the general framework of wetting theory, which generally describes the behavior of wetting layers forming on surfaces near phase transitions. Wetting transitions are theoretically well understood, and an important criterion for classification is the question if the transition is continuous or discontinuous. The corresponding experimental question here is, if the prefrozen film forms continuously or appears suddenly with a finite thickness. We here present an experiment on a polymeric model system, which allows a temperature dependent measurement of the prefreezing layer and shows that the transition is of first order.
In our experiment we use in-situ Atomic Force Microscopy measurements on an ultrathin film of a crystallizable polymer on graphite, which acts as a substrate inducing crystallization. Due to the general instability of the chosen sample system, it is possible for this case to directly measure the thickness of the prefrozen layer as a function of temperature.
Highlights 2017
Opposing Phase Segregation and Hydrogen-Bonding Forces in Supramolecular Polymers
Structure-formation in a wide range of materials and organisms is guided by self-assembly processes, leading to a high level of molecular organization, such as in folded macromolecules. Phase segregation between different macromolecules and specific weak interactions are the basis of molecular organization in many biological systems, held together by attractive hydrogen bonds (H-bonds),and distracted by phase segregation. We are reporting on significant changes in the association behaviour of the same, covalently attached H-bonds by the phase of attached polymer chains, revealed by a combination of synthetic methodologies, X-ray scattering and solid-state NMR-spectroscopy. Depending on the aggregation state we observe either closed H-bonds despite segregation of the phases, or macrophase separation with a larger amount of H-bonding dissociation. Thus the strength of a H-bond is significantly changed by the phase structure of the attached molecules such as polymers, displaying a phase segregating tendency.
Temperature-dependent IR-transition moment orientational analysis applied to thin supported films of poly-ε-caprolactone
When polymers crystallize from the melt, a huge variety of micro-structures can be obtained depending on the details of the production protocol. Despite about 100 years of application-driven and basic scientific research, the interplay of external parameters and microscopic mechanisms determining the micro-structure is still not unraveled. In this work, we analyze 7 µm thick, supported films of semi-crystalline Poly-ε-caprolactone using IR-Transition Moment Orientational Analysis. Based on the three-dimensional order of the crystalline and amorphous moieties, the films micro-structure is found to consist of extended flat on crystalline layers (~30%) and strongly confined spherulites. The former are commonly only found in nanometer-sized films, whereas the latter are usually dominant in such macroscopic samples. In the course of heating, the flat layers melt preferentially, whereas the spherulitic structures remain intact up to the equilibrium melting point. The findings are explained by the confined volume, accessible for crystal growth, nucleation kinetics, and the related thermodynamic stability. The work aids in understanding the interplay between kinetics and thermodynamics during the formation of polymer crystallites and their super-structures, and therefore helps to tailor polymeric materials for application.
- W. Kossack, M. Schulz, T. Thurn-Albrecht, J. Reinmuth, V. Skokow and F. Kremer, Soft Matter 13, 9211-9219 (2017)
See also
Induction of Chirality in β-Turn Mimetic Polymer Conjugates via Postpolymerization Click Coupling
Chirality is one of the most determining principles in chemistry and biology, dominating the assembly of large synthetic and biological macromolecules. Polymers with artificial (geometrical) constraints can - similar to alpha-helices or proteins - fold into well defined helical structures, especially those containing a rigid backbone with limited conformational flexibility. We here report on the transfer of chirality, induced by an artificial beta-turn-mimetic element, which is directly linked to a synthetic (helical) polymer. The distance over which chirality is transferred from the geometrical constraint onto the helicity of the polymers is studied, revealing that transfer of chirality is possible up to three chemical bonds between the helix and the chiral center on the beta-turn element.
Intracrystalline Jump Motion in Poly(ethylene oxide) Lamellae of Variable Thickness and the Underestimated Effect of Intracrystalline Chain Dynamics on the Morphology and Stability of Semicrystalline Polymers
It has long been known that the degree of crystallinity of linear polymers depends on the presence or absence of chain motion within and through the crystalline lamellae. While this fact has not yet found widespread appreciation or even explicit consideration in theoretical accounts of polymer crystallization, recent results from project A01 demonstrate that even the morphology is qualitatively different in the two cases. That is, polymers with intracrystalline mobility display lamellar stacks with well-defined amorphous-phase thickness but a wider distribution of crystallite thickness, in some contradiction to conventional wisdom. Polymers without αlphac-process on the other hand show a much better defined thickness of the crystalline regions. It is thus of high interest to develop an understanding of the relation between the intra-crystalline chain motion and morphological parameters, such as the crystallite thickness. Here, we report on a comprehensive NMR study of intracrystalline chain dynamics in poly(ethylene oxide) and a new approach for the quantitative analysis of small-angle X-ray scattering data. In the latter one, we compare the structural characteristics of fully crystallized samples for two model polymers with and without chain motion in the crystallites.
- R. Kurz, A. Achilles, W. Chen, M. Schäfer, A. Seidlitz, Y. Golitsyn, J. Kressler, W. Paul, G. Hempel, T. Miyoshi, T. Thurn-Albrecht, and K. Saalwächter, Macromolecules 50, 3890-3902 (2017)
See also
Highlights 2016
Spatial Orientation and Order of Structure-Defining Subunits in Thin Films of a High Mobility nType Copolymer
Orientation and order of distinct molecular subunits in solid layers of the high mobility n‑type copolymer P(NDI2OD‑T2) are investigated by means of infrared transition moment orientational analysis (IR‑TMOA). This novel spectroscopic technique based on concurrent absorbance measurements of structure-specific bands in dependence on inclination and polarization of the incoming IR light enables to determine the complete tensor of absorption independently for each IR-active transition moment. As a result, for nm‑thin films pronounced in‑plane anisotropy arising from self‑aggregated order is detected, which, however, is no longer discernable for mm-thick samples. In contrast, the out of plane orientation (inclination of molecular subunits) is retained irrespective of the widely varying layer thicknesses (150 nm vs. 1.4 mm). Thus, the conception of the sample morphology occurs as stratification of slightly misaligned layers of oriented polymers; with increasing film thickness the macroscopic in plain order diminishes, whereas the out of plain orientation is preserved.
- A. M. Anton, R. Steyrleuthner, W. Kossack, D. Neher, and F. Kremer, Macromolecules 49, 1798-1806 (2016)
See also
Dynamic Ordering and Phase Segregation in Hydrogen-Bonding Polymers
Hydrogen bonds constitute highly relevant structural units of molecular self-assembly. They bridge biological and synthetic sciences, implementing dynamic properties into materials and molecules. Phase segregation and crystallization on the other hand represent important assembly-principle, responsible for eg. cell compartimentation, membrane-formation and microphase segregation in polymers. We here discuss the phase segregation of H-bonding polymers in both, the solution and solid state, focusing on the specific aggregation of different H-bonding polymers in competition to phase segregation and crystallization. We illustrate that a rational architectural design within H-bonding polymer systems in interplay with phase segregation in both, the amorphous and crystalline state, opens perspectives to develop artificial supramolecular systems approaching the level of complexities and properties present in nature’s biomaterials.
Highlights 2015
Infrared Transition Moment Orientational Analysis on the Structural Organization of the Distinct Molecular Subunits in Thin Layers of a High Mobility n-Type Copolymer
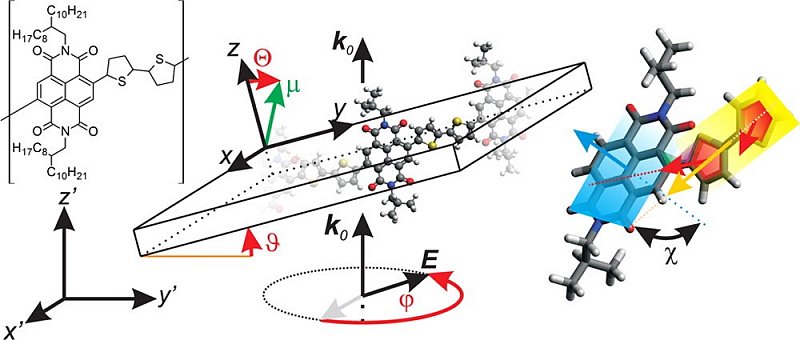
Organic electronics is the key technology for facing future energy challenges as large-scale devices can be manufactured at low costs using eco-friendly protocols. Organic solar cells, for instance, exhibit exceptional promising live cycle assessments, while organic light emitting diodes already became a commercial success. Since the morphology in a thin film influences its electronic structure and hence the device’s macroscopic properties, knowledge about the orientation and order of those molecules is highly demanding. Here we presented a suitable IR-based spectroscopic method and studied an n-type polymer. Solely on an experimental basis, it was possible to independently resolve the orientation of sub-molecular parts forming the polymer repeat unit and contribute to the fundamental understanding of structure formation.
Highlights 2014
Direct Observation of Prefreezing at the Interface Melt-Solid in Polymer Crystallization
The microscopic ordering process that a liquid undergoes during crystallization is often initiated at an interface to a solid. Different processes have been suggested by theory to occur at this interface. Of special interest is prefreezing—the formation of a thin crystalline layer at the interface already at temperatures above the melting temperature. Because of the difficult accessibility of the buried interface, experimental proof of crystallization by prefreezing has been elusive in molecular systems. We here present direct in situ observations of such a process in a polymeric model system. The results not only contribute to our fundamental understanding of crystallization but might also be useful for the preparation of well-ordered oriented thin films of crystalline organic materials.
Kinetic mechanism of chain folding in polymer crystallization
Polymer crystallization, which was discovered more than 50 years ago, is a particular case of solidification of matter which occurs by supercooling a polymer melt. The specific feature of polymer crystallization is the back-folding of chains in a polymer crystal leading to lamellae with a thickness of around 10-15 nm consisting of straight chain segments. The conventional description, which is based on phenomenological nucleation theory, ignores the polymeric structure and conformational interactions, and has been questioned in some recent experiments and numerical work. We develop a description of the crystallization kinetics that explains chain folding in polymer crystallization in terms of conformational interactions and the coil shape of polymer chains in the melt. The fundamental relation between lamellar thickness and supercooling is derived from the interplay between the formation time of rod shaped stems, which are induced by supercooling, and the relaxation time of coiled polymer parts of the same length. Our work suggests the existence of two separate time scales in polymer crystallization: the chain dynamics time scale at which the fold length is selected, and the much larger time scale associated with the crystal growth.
Highlights 2013
Determination of the Crystallinity of Semicrystalline Poly(3-Hexylthiophene) by Means of Wide-Angle X-Ray Scattering
Poly(3-hexylthiophene) (P3HT) is often studied as a model system for the more general class of semiconducting polymers. It has been shown in the past that the electronic transport properties of this material strongly correlate with crystallinity, which can depend on the details of chemical structure, molecular weight and processing. In most cases, crystallinity is measured by Differential Scanning Calorimetry (DSC), which requires calibration in order to deliver absolute values. Unfortunately, the available calibration value is uncertain and under debate. We therefore here present small-angle and wide-angle X-ray scattering measurements on a series of chemically well-defined poly(3-hexylthiophenes), which were analyzed to determine absolute values of the crystallinities. The resulting values for the crystallinity are substantially higher than assumed in the past. An extrapolated reference melting enthalpy for a 100% crystalline material was determined by comparison with DSC measurements. The observed decrease of the crystallinity for higher molecular weights can be explained by the onset of chain folding. Additionally we show that the crystalline regions of P3HT exhibit a large amount of internal disorder.
Crystallization of supramolecular pseudo block copolymers
Due to the presence of supramolecular bonds the crystallization process of supramolecular pseudo block copolymers (SPBCP) is more complex in comparison to conventional covalently bonded block copolymers (BCP). Thus supramolecular binding motives included on the polymer chain-ends display additional dynamic effects as well as possible nuclei for the crystallization. In this article we systematically study non-isothermal crystallization processes in SPBCPs consisting of a crystallizable poly(ε-caprolactone) (PCL) connected via triple hydrogen bonds to either a short alkyl-modified 2,4-diaminotriazine, or bound to a large block of amorphous poly(isobutylene) (PIB). The crystallization of the PCL is studied with both groups acting as supramolecular barriers for the crystallization process, either during nucleation or during crystal growth. A strong influence of the short alkyl-modified 2,4-diaminotriazine barrier on the crystallization temperature of the PCL compared to the control sample devoid of this compound is observed. In contrast, the large polymer block (PIB) acting as a barrier causes a strong decrease of the crystallization temperature and fractionated crystallization of SPBCP consisting of smaller PCL-chains is observed.
Hybrid systems
Highlights 2020
Hybrid polymers bearing oligo-l-lysine(carboxybenzyl)s: synthesis and investigations of secondary structure
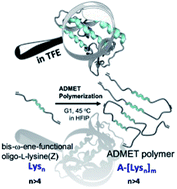
Hybrid polymers of peptides resembling (partially) folded protein structures are promising materials in biomedicine, especially in view of folding-interactions between different segments. In this study polymers bearing repetitive peptidic folding elements were successfully synthesized to study their secondary structure introduced by conformational interactions between their chains. We can also demonstrate the influence of chain length of the generated polymers on the formation of secondary structures.
Highlights 2019
Multisegmented hybrid-polymer based on oligo-amino acids: synthesis and secondary structure in solution and the solid state
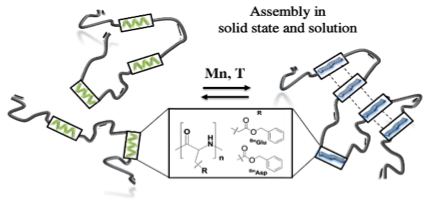
Proteins are among the most abundant macromolecules in nature, often forming complex architectures by folding (alpha-helices/beta-sheets) and/or aggregation into fibrillary assemblies. Refolding between different segments during assembly is often poorly understood, also guided by cooperative assembly phenomena. We here present a study on multisegmented hybrid-polymers to understand folding and assembly. Precisely engineered oligo-amino acids are repetitively imbedded into a non-interacting alkyl-chain, allowing to study conformational changes upon and during assembly in both, the solid state and in solution. The here reported observations prove that beta-sheets are thermodynamically favored, however strongly dependent on the nature of the amino-acid along the chain.
See also
Probing Polymer Chain Conformation and Fibril Formation of Peptide Conjugates
A collaborative study (projects A03/A06) investigated the conformation of hybrid molecules consisting of a thermo-responsive synthetic polymer and an amyloid-forming parathyroid hormone. Using NMR as a probe an attractive interaction between the two blocks leading to an intertwined shroud-like conformation of the synthetic polymer was found. Nevertheless, the synthetic block did not affect the secondary structure of the peptide and fibrillation was even accelerated in comparison to the pure peptide.
Highlights 2017
One-Pot Synthesis of Thermoresponsive Amyloidogenic Peptide-Polymer Conjugates via Thio-Bromo Click Reaction of RAFT Polymers
Conjugation to fibrillating proteins and peptides represents a methodological challenge, as potential binding sites are blocked due to the ongoing fiber-formation, blocking the sites for attachments. A synthetic strategy to efficiently prepare main-chain peptide-polymer conjugates developing an in situ tandem reaction based on the aminolysis/thio-bromo click reaction. This method allows to tether an amyloidogenic peptide fragment Aβ17-20(LVFF) to the ω-chain end of poly(diethylene glycol methyl ether acrylate) (PDEGA), prepared via reversible addition fragmentation chain transfer polymerization (RAFT). Tuning of the lower critical solution temperature of the polymer allows a modulation of the aggregation into micellar structures.